510(k) vs PMA vs De Novo: Which FDA Pathway Is Right for Your Device?
How to Choose the Right FDA Medical Device Approval Strategy - Before You Lose Time and Money
Bringing a product through medical device development is challenging enough—but choosing the wrong regulatory pathway can delay your timeline by months (or even years), increase costs, and jeopardize your chances of FDA medical device approval.
For companies navigating medical device design and medical device engineering, one of the most critical early decisions is selecting the correct FDA submission pathway:
510(k)
PMA (Premarket Approval)
De Novo
Each pathway has different requirements, timelines, risks, and strategic implications.
In this guide, we’ll break down the differences between these FDA pathways, when to use each, and how to align your medical device product development strategy for faster, smoother approval.

What Are FDA Medical Device Approval Pathways?
FDA medical device approval pathways are regulatory routes that determine how a device is evaluated before it can be marketed in the United States.
These pathways are based on:
Device risk classification (Class I, II, III)
Novelty of the technology
Availability of predicate devices
Intended use and user risk
The Three Primary FDA Pathways:
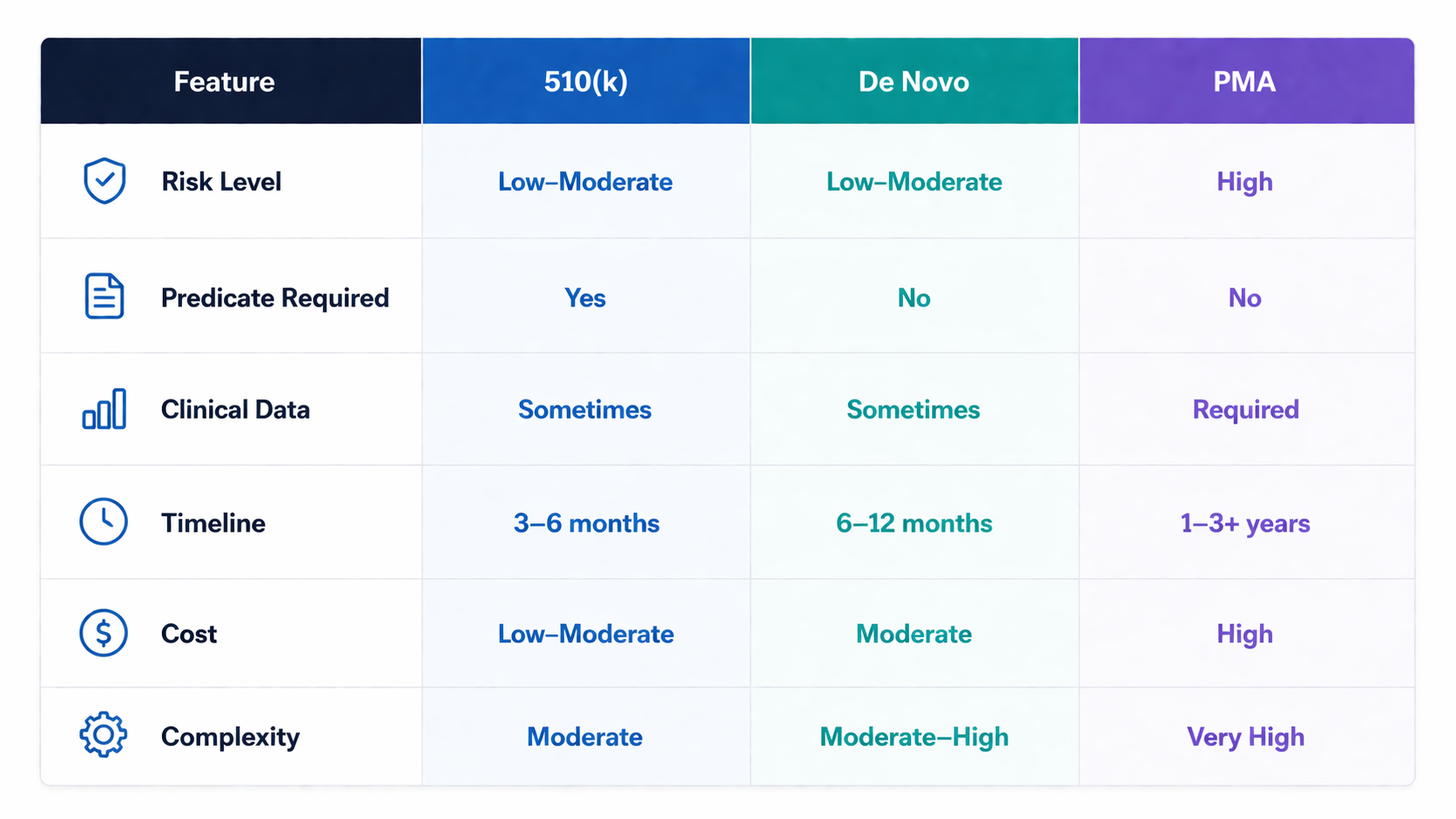
510(k): Demonstrates substantial equivalence to an existing device
De Novo: For novel, low-to-moderate risk devices with no predicate
PMA: Required for high-risk devices with significant safety concerns
Choosing the wrong pathway can lead to:
Rejected submissions
Additional testing requirements
Significant development delays
510(k) Pathway: The Most Common Route
What Is a 510(k)?
A 510(k) submission demonstrates that your device is “substantially equivalent” to a legally marketed predicate device.
This is the most common pathway for medical device development companies because it is generally faster and less expensive than PMA.
When to Use 510(k)
Use this pathway if:
A similar device already exists on the market
Your device has the same intended use
Differences do not raise new safety or effectiveness concerns
Key Requirements:
Predicate device comparison
Performance testing (bench, software, etc.)
Verification and validation medical devices data
Labeling and intended use documentation
Typical Timeline:
3–6 months (can extend depending on FDA questions)
Common Pitfalls:
Choosing an inappropriate predicate
Insufficient testing data
Poorly defined intended use
Strategic Insight:
Many companies underestimate how critical early medical device design decisions are to 510(k) success. Small design differences can trigger additional regulatory scrutiny.

De Novo Pathway: For Novel, Lower-Risk Devices
What Is De Novo?
The De Novo pathway is designed for devices that are:
New (no predicate exists)
Low to moderate risk
It allows the FDA to create a new device classification.
When to Use De Novo:
Your device is innovative
No substantially equivalent product exists
Risk profile does not require PMA
Key Requirements:
Full description of device and intended use
Risk analysis (ISO 14971 aligned)
Performance data
Human factors engineering medical devices validation (if applicable)
Typical Timeline:
6–12 months
Advantages:
Creates a new classification (future competitors can use 510(k))
Less burdensome than PMA
Challenges:
More uncertainty
Greater documentation expectations than 510(k)
Strategic Insight:
De Novo is often misunderstood. Companies sometimes attempt a 510(k) first—only to be rejected and forced into De Novo, losing valuable time.

PMA (Premarket Approval): The Most Rigorous Pathway
What Is PMA?
Premarket Approval (PMA) is required for Class III devices—those that:
Sustain or support life
Are implanted
Present significant risk
This is the most stringent type of FDA medical device approval.
When to Use PMA:
No predicate exists
Device is high risk
Clinical data is required
Key Requirements:
Extensive clinical trials
Scientific evidence of safety and effectiveness
Manufacturing process validation
Full FDA compliance medical devices documentation
Typical Timeline:
1–3+ years
Challenges:
High cost (often millions of dollars)
Long timelines
Complex regulatory interactions
Strategic Insight:
PMA success is heavily dependent on early-stage medical device engineering, study design, and regulatory alignment. Mistakes early in development are extremely costly to fix later.
510(k) vs De Novo vs PMA: Key Differences:
How to Choose the Right FDA Pathway
Selecting the correct pathway isn’t just a regulatory decision—it’s a business-critical strategy that impacts your entire medical device development process.
Key Questions to Ask:
1. Does a Predicate Device Exist?
If yes → 510(k) may be viable
If no → consider De Novo or PMA
2. What Is the Risk Classification?
Class I/II → 510(k) or De Novo
Class III → PMA
3. How Novel Is the Technology?
Incremental innovation → 510(k)
Breakthrough innovation → De Novo or PMA
4. What Level of Evidence Is Required?
Bench testing only → 510(k)
Clinical trials → PMA (and sometimes De Novo)
The Hidden Risk: Choosing the Wrong Pathway
One of the most common—and costly—mistakes in medical device product development is pursuing the wrong regulatory pathway.
What Happens When You Get It Wrong:
FDA rejection or “Refuse to Accept” (RTA)
Additional testing requirements
Redesign of your device
Delayed market entry
Real-World Scenario:
A company develops a device assuming a 510(k) pathway. Midway through submission, the FDA determines:
No valid predicate exists
Result:
Forced into De Novo
6–12 month delay
Increased costs
Why Regulatory Strategy Must Start Early
Regulatory planning should begin during the earliest stages of medical device design—not just before submission.
Early Integration Impacts:
Product requirements
Risk management strategy
Human factors testing medical devices
Verification and validation plans
Design controls medical devices compliance
FDA Expectation:
Under 21 CFR Part 820, design controls must be integrated throughout development—not added later.
The Role of Human Factors and Usability in FDA Approval
Regardless of pathway, human factors engineering medical devices is increasingly critical.
Why It Matters:
Reduces use errors
Improves patient safety
Supports FDA approval
FDA Focus:
Devices used in:
Home environments
High-risk scenarios
Complex workflows
Often require:
Formal usability testing
Human factors validation studies
Ignoring this early can:
Trigger additional FDA requirements
Cause redesigns and delays

How an End-to-End Partner Reduces Regulatory Risk
Navigating FDA pathways requires more than regulatory knowledge—it requires alignment across:
Medical device design
Engineering
Human factors
Testing
Regulatory strategy
The Challenge with Fragmented Teams:
Misaligned assumptions
Communication gaps
Delays in decision-making
The Advantage of an Integrated Device Development Partner Like HaA PD:
A full-service medical device development company like HaaPD provides:
FDA consulting for medical devices
Regulatory pathway strategy
Medical device prototyping services
Human factors engineering
UI/UX design for medical devices
Verification and validation support
This integrated approach ensures:
Regulatory strategy is aligned from day one
Fewer surprises during submission
Faster time to market
Key Takeaways
Choosing between 510(k), De Novo, and PMA is one of the most critical decisions in medical device development
The right pathway depends on risk, novelty, and available predicates
Early regulatory strategy directly impacts timeline, cost, and success
Human factors, design controls, and testing must be integrated from the start
Working with an experienced partner significantly reduces risk and accelerates FDA approval
Ready to Navigate FDA Approval with Confidence?
Whether you’re early in medical device design or preparing for submission, selecting the right FDA pathway is critical to your success.
HaaPD helps companies:
Define the optimal regulatory strategy
Align design and engineering with FDA expectations
Reduce delays and accelerate approval