Medical Device Development Process: From Concept to FDA Approval
How to Bring a Medical Device to Market Faster—Without Costly Mistakes
Bringing a medical device to market is a complex, highly regulated process that requires more than just a great idea. From early concept development to FDA approval, every stage must be carefully executed to ensure safety, compliance, and commercial success.
Whether you’re a startup founder or part of an established MedTech company, understanding the medical device development process can help you avoid delays, reduce costs, and accelerate time to market.
In this guide, we’ll break down each phase—and show how an experienced medical device development partner like HaA PD can help you succeed.
What Is the Medical Device Development Process?
The medical device development process is a structured pathway that takes a product from concept through design, engineering, testing, and regulatory approval.
It includes:
Product strategy and concept development
Design controls and engineering
Human factors and usability testing
Verification and validation
FDA submission and clearance
Manufacturing and commercialization
Each phase is critical—and skipping steps or making errors can lead to costly delays or regulatory rejection.
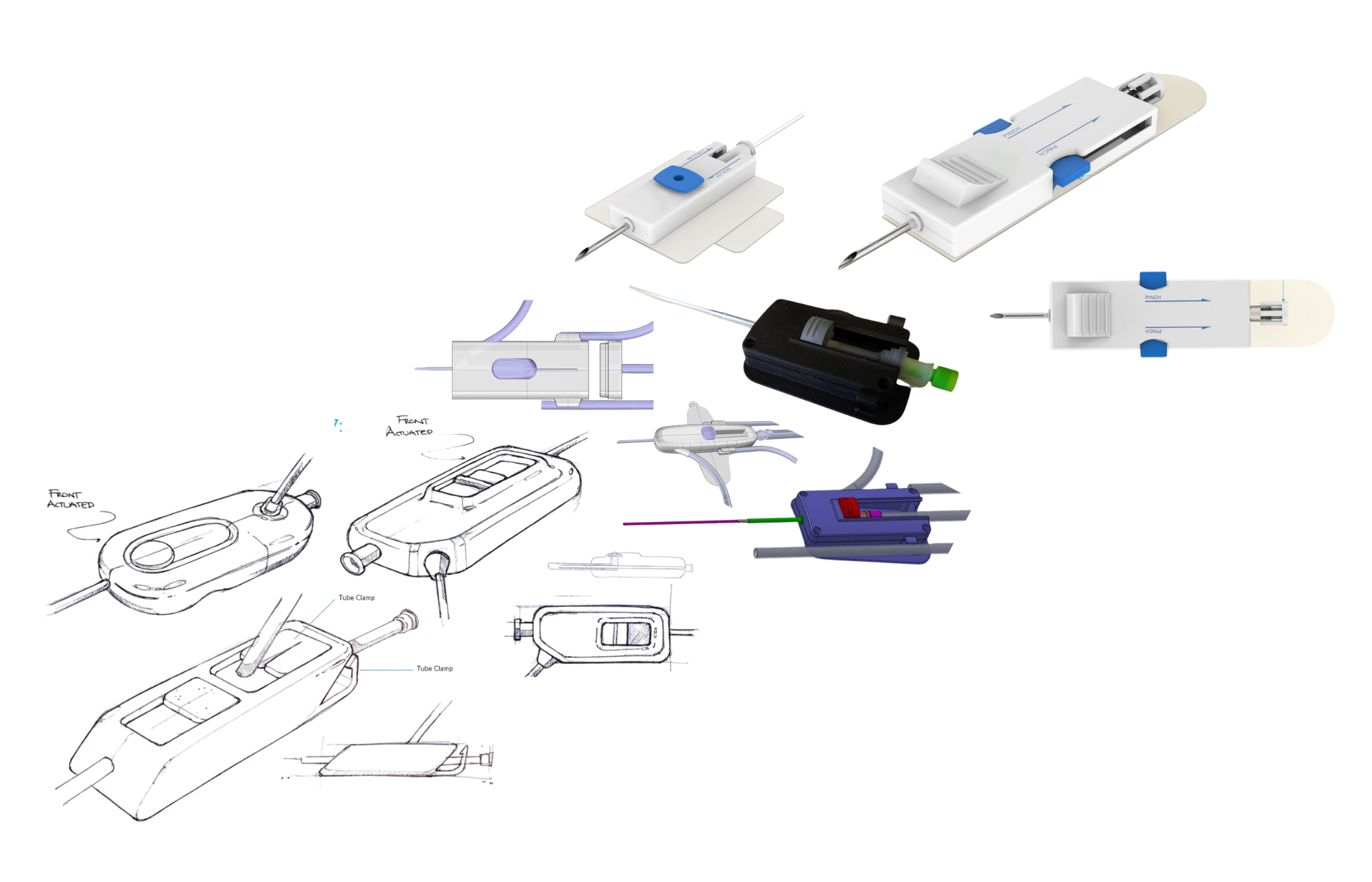
Step 1: Concept Development and Feasibility
Every successful medical device starts with a clear understanding of:
The clinical need
The target user
The regulatory pathway
At this stage, teams define:
Product requirements
Intended use
Risk profile
Why this matters:
Poorly defined requirements early on are one of the leading causes of FDA delays.
This is also where early human factors engineering should begin—ensuring the product is intuitive and safe for real users.
Step 2: Design Controls and Product Development
FDA design controls (required under 21 CFR 820) ensure that medical devices are developed systematically and safely.
This phase includes:
User needs → design inputs
Design outputs (specifications, drawings)
Risk management (ISO 14971)
Iterative prototyping
Step 3: Human Factors Engineering and Usability Testing
Human factors engineering is no longer optional—it’s a critical requirement for FDA approval.
It ensures:
Devices are safe and intuitive
Use errors are minimized
Real-world conditions are accounted for
FDA Focus:
The FDA requires human factors validation testing for many devices, especially those used in home or high-risk environments.
Common mistake: Companies often treat usability as a late-stage activity—this can lead to redesigns and major delays.
Step 4: Engineering, Prototyping, and Iteration
During this phase, the product evolves from concept to a functional device.
Key activities include:
Mechanical, electrical, and software engineering
Rapid prototyping
Design for manufacturability (DFM)
Why design for manufacturability matters:
If manufacturing constraints aren’t considered early:
Costs increase
Timelines slip
Quality issues emerge
Step 5: Verification and Validation (V&V)
Verification and validation are essential for proving that:
The device meets design specifications (verification)
The device meets user needs (validation)
In simple terms:
Verification: “Did we build it right?”
Validation: “Did we build the right thing?”
Includes:
Bench testing
Software validation
Biocompatibility testing
Human factors validation
Step 6: FDA Submission and Approval
The regulatory pathway depends on the device classification:
510(k): Substantial equivalence to an existing device
De Novo: New, low-to-moderate risk devices
PMA: High-risk devices requiring clinical data
Common pitfalls:
Incomplete documentation
Poor risk analysis
Inadequate testing data
Step 7: Manufacturing and Commercialization
Once FDA clearance is achieved, the focus shifts to:
Scaling production
Quality systems (ISO 13485)
Supply chain management
Critical factor:
Devices that are not designed for manufacturing early often face:
Production delays
Cost overruns
Quality issues
Why Many Medical Device Projects Fail (and How to Avoid It)
The most common reasons for delays include:
Lack of regulatory strategy early on
Poor integration between design and engineering
Ignoring human factors until late stages
Inadequate verification and validation
The solution:
Work with a partner that integrates:
Design
Engineering
Human factors
Regulatory strategy

The Advantage of an End-to-End Medical Device Development Partner
Managing multiple vendors across design, engineering, testing, and regulatory can lead to:
Miscommunication
Delays
Increased costs
An integrated partner like HaA PD streamlines the entire process by providing:
Human factors engineering
UI/UX design for medical devices
Full-stack engineering
FDA consulting and regulatory strategy
This unified approach reduces risk and accelerates development timelines.
Key Takeaways
The medical device development process is complex—but manageable with the right strategy
Early decisions around design, usability, and regulatory pathways have the biggest impact
Human factors and FDA compliance must be integrated from the start
An experienced development partner can significantly reduce time to market
Ready to Accelerate Your Medical Device Development?
If you’re developing a new medical device—or looking to bring an existing concept to market—HaA PD can help you navigate every stage of the process.