What is a Class III Medical Device? | HaA PD
What You Will Learn in This Article:
How the FDA classifies medical devices into Class I, II, and III based on risk level.
What defines a Class III medical device and why it represents the highest-risk category.
The regulatory requirements for Class III devices, including Premarket Approval (PMA) and clinical evidence.
The role of design controls and clinical validation in securing product safety, quality, and compliance.
Examples of Class III medical devices developed with expert support.
Introduction to Medical Device Classification

The United States Food and Drug Administration (FDA) categorize medical devices into three different classes, depending on the level of regulatory control required to maintain both performance and safety. The FDA classifies Class III devices as medical devices that pose the highest risk to both users and patients, requiring the most stringent regulatory oversight.
Class III devices are considered higher risk than both Class I and Class II devices due to their intended use and potential impact on patient health. These devices are often life-sustaining, life-supporting, implanted, or critical to preventing serious injury or illness.
While Class III devices represent a smaller percentage of all medical devices, they require significantly more regulatory scrutiny, clinical evidence, and development discipline than class I and II devices.
Image to left: Bone Screw
How Are Medical Devices Classified in the US?
The three classes defined by the US Food and Drug Administration (FDA) for the United States medical device market range from Class I to Class III. This classification system is based on the progressive risk to the user or patient.
Class I devices pose the lowest risk, and Class III devices pose the highest risk, with each classification level requiring progressively more stringent regulatory control. This risk-based approach allows the FDA to implement controls that are proportional to the potential harm posed by the device.
Most medical devices can be classified by determining the corresponding description of the device in the specialty “panels” of Title 21 of the CFR. Title 21 of the Code of Federal Regulations (CFR) contains U.S. federal regulations for products regulated by the FDA and the DEA (Drug Enforcement Administration).
Class I
Class I devices pose the lowest risk and are typically subject to general and straightforward controls, including registration, listing, and labeling requirements. Examples of Class I devices include bandages, plasters, medical spoons, glasses, and stethoscopes. The majority of these devices are exempt from the requirement for premarket notification.
Class II
Class II devices pose a slightly higher to moderate risk and therefore require general controls and some special controls, which may include performance standards, special labels, or post-market surveillance. Examples are contact lenses, biopsy forceps, hearing aids, and infusion pumps. The majority of these devices require a premarket notification, which is a submission made to the FDA to show that the device is substantially equivalent (as safe and effective as a device already legally marketed and available on the market
Class III
Class III devices have the highest risk factor and need general controls as well as Premarket Approval (PMA). This type of device is typically life-sustaining or life-supporting, implanted, or presents a potential risk of serious injury or illness if they fail.
Examples include pacemakers, defibrillators, cochlear implants, heart valves, and neurostimulation systems.
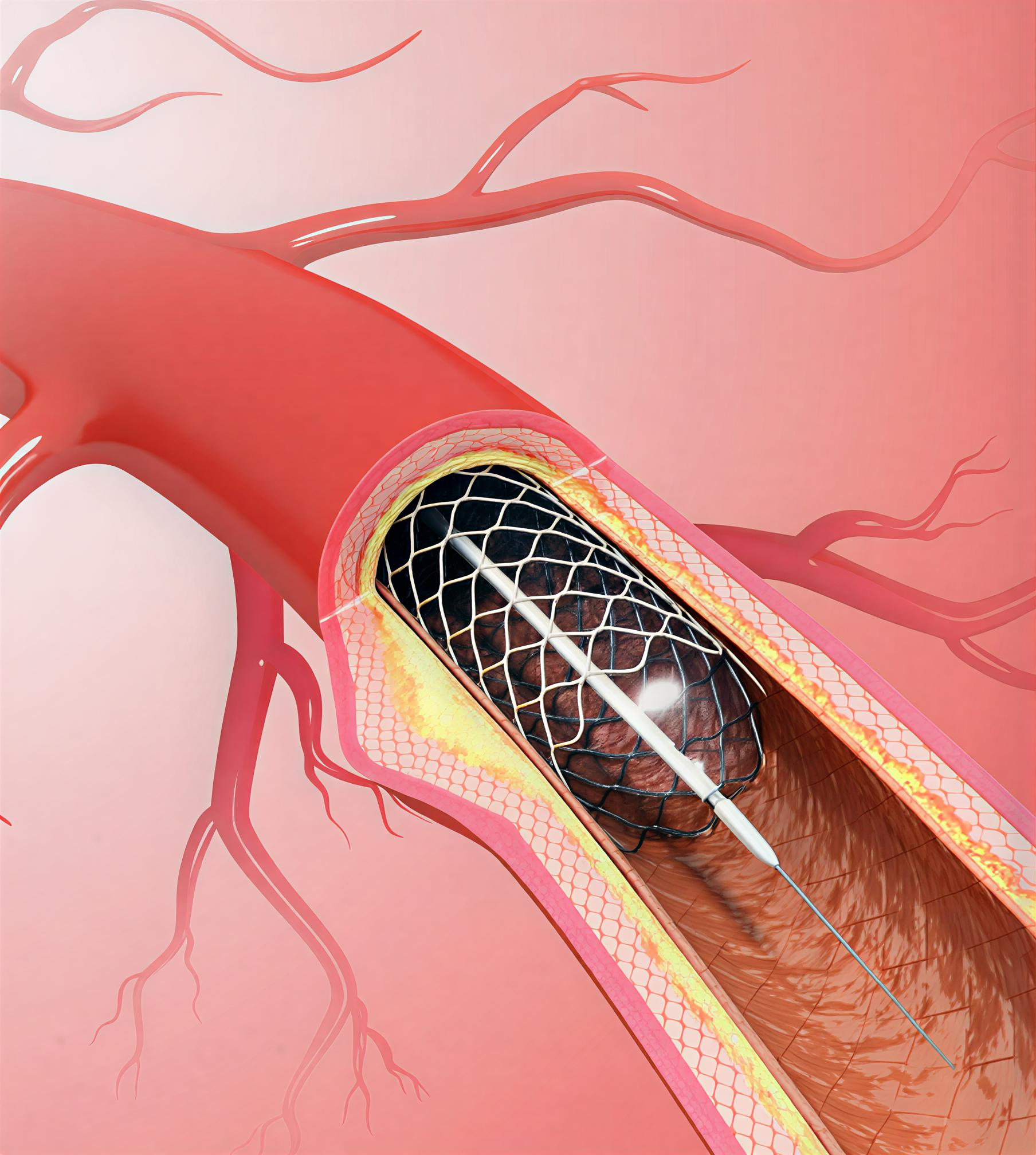
What Makes a Medical Device Class III?
Class III medical devices are the most complex products regulated by the FDA. These devices require a higher level of proof of safety and effectiveness than Class I or Class II devices.
As a device developer, manufacturer or medical professional, it is important to understand what constitutes a Class III medical device. These devices often interact directly with critical physiological systems, such as the heart or brain, and remain in the body for extended periods of time. Their characteristics usually require the following:
Premarket Approval (PMA)
For Class III devices, this means obtaining Premarket Approval (PMA), which requires independent scientific evidence demonstrating that the device is safe and effective for its intended use.
Clinical Evidence
Most Class III devices require clinical studies, prior to and as a prerequisite for, PMA approval, which are conducted under an Investigational Device Exemption (IDE).
Example of a Class III Medical Device Developed by HaA PD
At HaA PD, we provide medical device development services from concept and design through testing, clinical readiness, and manufacturing. We have supported Class III medical device programs that require strict adherence to FDA design controls and PMA-level documentation. Examples of Class III medical devices and systems HaA PD has supported include heart stents, bone anchors, bone screws, knee joints, hip joints and defibrillators. These programs demand disciplined design controls, risk management, and close coordination between engineering, clinical, and regulatory teams.
FDA Requirements for Class III Devices
FDA Class III devices require strict regulation to ensure device effectiveness and the highest level of patient safety. These devices are subject to general controls and must obtain Premarket Approval (PMA) before being marketed in the United States.
PMA approval requires manufacturers to submit extensive documentation, including device design details, nonclinical testing, clinical study data, labeling, and manufacturing information. The FDA conducts a comprehensive scientific review to confirm that the benefits of the device outweigh the associated risks.
Class III device manufacturers must comply with the Quality Management System Regulation (QMSR), which governs all aspects of design, manufacturing, packaging, labeling, record-keeping, and product traceability. The QMSR incorporates ISO 13485:2016 by reference, which is the international standard that specifies the requirements for a quality management system (QMS), which Class III device manufacturers are required to develop and maintain. Design control is a critical component of QMSR compliance and requires manufacturers to document, validate, and manage the design process to ensure safety, quality, and regulatory compliance.
Failure to comply with FDA regulations can result in enforcement actions including warning letters, recalls, civil penalties, or withdrawal of PMA approval.
The Approval Process of Class III Devices
FDA approval for a Class III medical device is a structured, multi-step process that requires early planning from the start of development and a high level of regulatory and quality standards expertise.
Step 1: Classify your device
The first step is to classify your device using the FDA Product Classification Database. This process requires you to define your intended use and identify the medical specialty it best suits. Medical devices that are implantable, life-sustaining, or novel in nature are often classified as Class III.
Step 2: Establish a regulatory strategy
The second part of the process is to develop and plan a regulatory strategy that includes comprehensive, compliant study planning to collect clinical data. As a prerequisite to approval, most Class III devices require an Investigational Device Exemption (IDE) to conduct clinical studies to demonstrate safety and effectiveness. In addition, it is critical at this step to implement a Quality Management System compliant with ISO13485:2016.. In this step, it is important you align with a team of experts in the areas of regulatory, quality and clinical study planning. Missed steps, or information can cost you your approval and / or significantly delay the process, costing you time and money.
Step 3: Conduct clinical studies
The third step is to execute your clinical study to generate valid scientific evidence supporting safety and effectiveness of your medical device. These studies and the data collected will form the foundation of your PMA submission.
Step 4: Submit your PMA
Once the design, testing, and clinical studies are complete, you will submit a PMA application to the FDA. Upon review, the FDA may issue deficiencies during review, which must be addressed promptly. It is critical to engage with experts to avoid delays and potential stop-gates.
Step 5: Approval and post-market requirements
After receiving PMA approval, manufacturers must complete additional processes and steps to complete the process. These three critical regulatory requirements include:
Establishment Registration: Manufacturers, and often importers or specification developers, must register their facility with the FDA annually between October 1 and December 31.
Device Listing: Manufacturers must list their device and its essential information in the FDA's database, which links the approved product to the registered facility.
UDI Compliance (Unique Device Identification): Manufacturers must assign a unique identifier to the device and its packaging and submit this information to the FDA’s Global Unique Device Identification Database (GUDID).
In addition, for the life of the device manufacturers must comply with FDA-governed post-market surveillance requirements to monitor safety, investigate complaints, file Medical Device Reports (MDRs), and manage corrections/removals.
These actions ensure that after a medical device is PMA approved, the product and its manufacturing site are traceable, properly identified, and compliant with ongoing FDA oversight, including potential inspections.
Take the Next Step Toward FDA Approval
Bringing a Class III medical device to market requires more than innovation. It requires a disciplined development process, strong clinical evidence, and expert regulatory execution. HaA PD has years of experience working with the complex processes, requirements and systems associated with PMA medical device approval. HaA PD partners with medical device developers to navigate the complexity of Class III development with confidence.
Key Takeaways:
Class III medical devices pose the highest risk and require Premarket Approval (PMA).
Most Class III devices require clinical studies conducted under an Investigational Device Exemption (IDE).
Manufacturers must comply fully with stringent FDA regulations such as the Quality Management System Regulation (QMSR), including strict design controls.
Examples of Class III medical devices are pacemakers, heart valves, cochlear implants, and neurostimulation devices.
Expert guidance is essential to manage regulatory complexity, and reduce risk.