Top 10 Mistakes That Delay Medical Device FDA Clearance
How to Avoid Regulatory Setbacks, Reduce Risk, and Accelerate Medical Device Product Development
Bringing a medical device to market is a complex process that requires far more than a great idea and strong engineering.
Even highly innovative products can face significant delays when companies underestimate the regulatory requirements needed for FDA clearance.
For many organizations, the biggest obstacle isn't the technology itself-it's the development process.
Missed requirements, weak documentation, inadequate testing, and poor regulatory planning can all trigger additional FDA questions, lengthy review cycles, and costly redesigns.
For companies investing in medical device product development, medical device design, and medical device engineering, understanding the most common mistakes that delay FDA clearance is essential for reducing risk, controlling costs, and accelerating time to market.
In this guide, we'll examine the top 10 mistakes that frequently delay FDA clearance and explore strategies that can help organizations navigate the medical device development process more efficiently.
Why FDA Clearance Delays Happen
Many companies assume that FDA clearance delays occur because their technology doesn't work.
In reality, delays are often caused by process failures rather than technical failures.
Common issues include:
Incomplete regulatory strategies
Insufficient design controls
Poor risk management documentation
Inadequate verification and validation testing
Human factors deficiencies
Weak FDA submission packages
The FDA's primary responsibility is protecting patient safety.
When reviewers cannot clearly determine that a device is safe and effective, they request additional information-which can significantly extend review timelines.
Strategic Insight
FDA clearance is often determined long before submission. The quality of your medical device development process directly impacts the speed of FDA review.
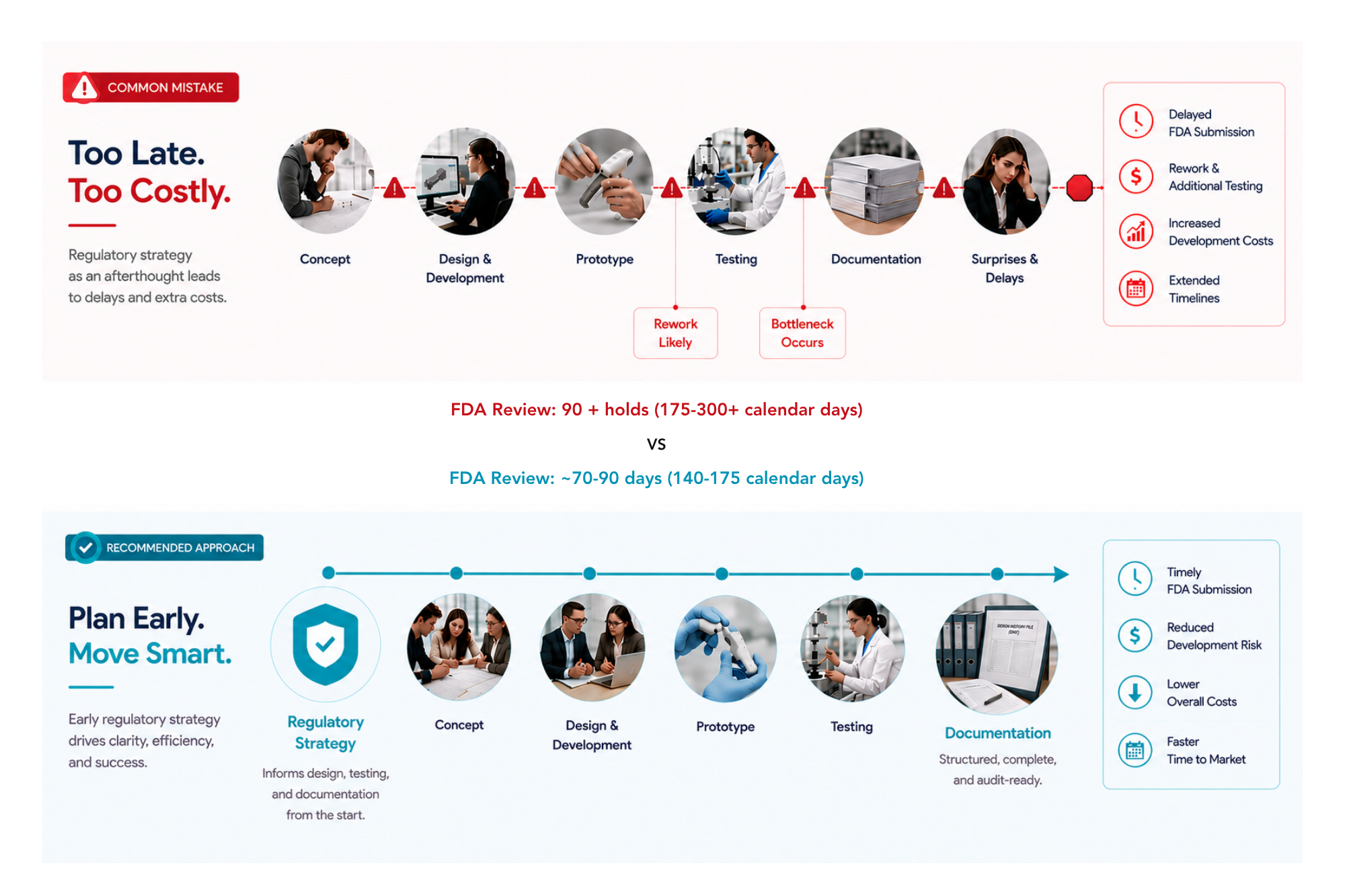
Mistake #1: Waiting Too Long to Develop a Regulatory Strategy
One of the most common causes of delay is treating regulatory planning as an afterthought.
Many companies focus heavily on engineering and product development before fully understanding their regulatory pathway.
Questions that should be answered early include:
Will the device require a 510(k) submission?
Is there an appropriate predicate device?
Could the device require De Novo classification?
What testing requirements will apply?
Without a clear regulatory strategy, development teams often discover late-stage gaps that require additional testing or documentation.
Impact:
Delayed FDA submissions
Additional development costs
Extended timelines
Best Practice
Integrate regulatory strategy into the earliest stages of medical device product development.

Mistake #2: Poorly Defined Design Controls
FDA design controls are foundational to successful medical device development.
Under FDA regulations, manufacturers must establish and maintain documented design controls throughout development.
These controls help demonstrate that:
User needs have been identified
Design inputs are properly defined
Design outputs meet requirements
Verification and validation activities have been completed
Common Problems:
Missing design inputs
Weak requirements documentation
Incomplete traceability
Poor design review records
Impact:
FDA questions during review; documentation deficiencies; potential resubmission requirements.

Mistake #3: Inadequate Risk Management
Risk management is not simply a regulatory requirement-it is a critical component of medical device safety.
FDA reviewers expect risk management activities to align with ISO 14971 throughout the development process.
Common deficiencies include:
Incomplete hazard analysis
Failure to identify use-related risks
Weak mitigation strategies
Poor risk traceability
The FDA expects clear evidence showing that identified risks have been addressed through design, testing, or other controls.
Impact:
Additional FDA review cycles
Requests for clarification
Delayed clearance decisions

Mistake #4: Treating Human Factors Engineering as a Final Step
Many organizations delay human factors engineering until validation testing.
This is one of the most expensive mistakes a company can make.
Human factors engineering medical devices should be integrated throughout development to ensure users can safely and effectively interact with the device.
The FDA increasingly scrutinizes:
User interfaces
Instructions for use
Home-use environments
Critical task performance
Common Consequences:
Failed usability validation studies
Device redesigns
Repeat testing
Best Practice
Conduct formative usability testing early and often. The sooner usability issues are identified, the easier they are to fix.

Mistake #5: Weak Verification and Validation Planning
Verification and validation medical devices activities are essential for demonstrating that a device meets its requirements and intended use.
Verification Answers:
Did we build the device correctly?
Validation Answers:
Did we build the correct device?
Common Issues:
Missing test protocols
Inadequate sample sizes
Incomplete test coverage
Poor documentation
Impact:
FDA requests for additional testing; delayed submissions; increased development costs.

Mistake #6: Insufficient Medical Device Testing
Testing deficiencies are among the most frequent reasons for FDA clearance delays.
Depending on the device, testing requirements may include:
Electrical safety testing
Biocompatibility testing
Software validation
Sterilization validation
Packaging validation
Shelf-life testing
Performance testing
Many companies underestimate the time required to complete testing programs.
Strategic Insight
Testing delays often create cascading delays throughout the entire medical device development process.
Planning early is critical.

Mistake #7: Poor Documentation and Design History File Management
If it isn't documented, the FDA assumes it didn't happen.
A complete Design History File (DHF) demonstrates that development activities were conducted according to regulatory requirements.
Common Documentation Problems:
Missing records
Inconsistent documentation
Poor traceability
Incomplete design reviews
FDA reviewers frequently identify documentation gaps that trigger requests for additional information.
Impact:
Lengthy review cycles
Increased regulatory risk
Potential submission rejection

Mistake #8: Overlooking Medical Device UI/UX Design
As medical devices become increasingly software-driven, medical device UI UX design has become a major regulatory focus.
Poor interface design can contribute to:
User confusion
Incorrect operation
Increased cognitive load
Safety risks
Strong UI/UX design improves:
User adoption
Training efficiency
Safety outcomes
Product performance
Best Practice
Incorporate UI/UX expertise early in medical device design and development.

Mistake #9: Failing to Align Engineering and Regulatory Teams
Many delays occur because engineering, quality, and regulatory teams operate in separate silos.
When teams are not aligned:
Design decisions may create regulatory challenges
Testing plans may miss critical requirements
Documentation may become inconsistent
Successful organizations establish cross-functional collaboration throughout development.
This alignment ensures:
Consistent decision-making
Better traceability
Faster issue resolution
Result:
Reduced risk and more efficient FDA submissions.

Mistake #10: Choosing the Wrong Development Partner
Medical device development requires expertise across multiple disciplines.
Organizations often struggle when they rely on partners who specialize in only one aspect of development.
Successful FDA clearance requires integration of:
Medical device design
Medical device engineering
Human factors engineering
Verification and validation
Regulatory strategy
Quality systems
Without this integrated approach, communication gaps and missed requirements can create significant delays.
Strategic Insight
An experienced medical device development partner can identify risks early, streamline development activities, and improve regulatory outcomes.
The Cost of FDA Clearance Delays
Even a relatively short delay can have significant business consequences.
Potential impacts include:
Lost revenue opportunities
Increased development costs
Delayed market entry
Competitive disadvantages
Additional testing expenses
Resource constraints
For startup and growth-stage companies, delays can also impact funding timelines and commercialization plans.
How Early Planning Accelerates FDA Clearance
Organizations that consistently achieve efficient FDA clearance timelines typically focus on:
Early regulatory strategy development
Strong design controls
Comprehensive risk management
Human factors engineering integration
Thorough verification and validation planning
Complete documentation
Cross-functional collaboration
These activities reduce uncertainty and improve submission quality.
The result is a smoother path through FDA review.
How HaA PD Helps Reduce FDA Clearance Delays
As a full-service medical device development company, HaA PD helps organizations navigate complex regulatory requirements while minimizing risk throughout the development process.
HaA PD Capabilities Include:
Medical device product development
Medical device design and engineering
Regulatory strategy development
FDA 510(k) support
Human factors engineering
Medical device usability testing
Risk management
Verification and validation support
FDA consulting for medical devices
Design controls implementation
The HaA PD Advantage
HaA PD aligns:
Product strategy
Engineering
Quality systems
Human factors
Regulatory requirements
Result:
Reduced development risk
Stronger FDA submissions
Faster approval timelines
More efficient commercialization
Key Takeaways
FDA clearance delays are often caused by process issues rather than technical failures
Early regulatory strategy is critical for successful medical device development
Design controls and risk management must be integrated throughout development
Human factors engineering plays a major role in FDA approval outcomes
Verification, validation, and testing deficiencies frequently trigger delays
Strong documentation and cross-functional collaboration improve regulatory success
An experienced medical device development partner can significantly reduce risk and accelerate timelines
Ready to Accelerate FDA Clearance?
If you're developing a medical device, avoiding common regulatory pitfalls can dramatically improve your path to market.
HaA PD helps companies:
Build effective regulatory strategies
Reduce development risk
Strengthen FDA submissions
Accelerate medical device FDA clearance
> Contact HaA PD today to learn how our integrated medical device product development expertise can help bring your innovation to market faster.